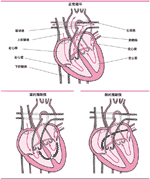
| . 房间隔、室间隔缺损
房间隔和室间隔缺损是指把心脏分成左右两半的间隔存在一个异常通道。
房间隔缺损是发生在心脏上部分的腔之间(心房)的缺损,心房接受回到心脏的血液。室间隔缺损是发生在心脏下部分的腔之间(心室)的缺损,心室将血液泵出心脏。
房间隔、室间隔缺损时,血液在肺和心脏间形成了一个短的循环。左心室收缩时,血液流到肺部,而没有泵到全身的其他部位,肺血管内血流量增加,出现呼吸短促、喂养困难、多汗、体重不增等。这些症状在室间隔缺损的儿童较常见。房间隔缺损,一般在小孩1岁以后才被发现,房间隔缺损小儿症状不明显。
有些儿童除心电图、心动图和X线胸片检查外,还需作心导管检查以明确诊断。房间隔、室间隔缺损都可以手术修复。
. 动脉导管未闭
动脉导管未闭是指主动脉(携带动脉血到全身的大动脉)和肺动脉(携带静脉血到肺的动脉)之间存在异常通道。
动脉导管可以使动脉血分流到肺,在胎儿期,这个功能是存在的,由于胎儿不能呼吸空气,因此血液不需要流经肺进行气体交换。但出生后,血液必须流到肺进行气体交换。正常情况下,动脉导管在生后1~2天很快闭合。如果动脉导管未闭,流到全身的动脉血又回到肺,肺循环血流量增加,婴儿可能出现心力衰竭,其症状为呼吸困难、心率快和体重不增。
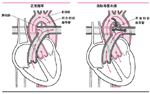
动脉导管未闭的足月新生儿可能在生后几周内出现心力衰竭。因此,必须手术关闭动脉导管。听诊器听到动脉导管未闭的杂音,常可确定动脉导管未闭。能听到杂音的小儿一般在1岁左右择期手术,以防止严重感染。
动脉导管未闭早产儿较足月儿常见,早产儿肺循环血液量增加,可能严重影响心脏和未成熟的肺功能。动脉导管未闭的早产儿,应限制输液量,并使用消炎痛药物治疗或进行外科手术。
. 主动脉瓣狭窄
主动脉瓣狭窄是指主动脉瓣膜的狭窄,主动脉瓣开放时左心室将血泵到主动脉,然后到全身(见第19节)。
主动脉瓣正常由三个叶片(瓣膜)组成,瓣膜关闭或开放让血流通过。主动脉瓣狭窄患者主动脉瓣只有两叶,导致开放不全,限制了血流量。左心室必须用更大的压力将血泵出瓣膜口。有些主动脉狭窄的儿童病情严重,需要外科手术矫正。成年人的主动脉瓣狭窄更常见。有些小儿出现心力衰竭,全身血流量减少,出现缺血缺氧。出现心力衰竭必须急诊治疗,治疗包括药物治疗,急诊外科手术,或作气囊瓣膜成形术。气囊瓣膜成形术是用一个带气囊倾斜的管子(导管)将瓣膜扩张并撕裂。在年长儿或青少年,瓣膜可以用外科手术切开或用人工瓣膜替换,根据病情选择不同的治疗方法。
. 肺动脉瓣狭窄
肺动脉瓣狭窄是指肺动脉瓣膜的狭窄,肺动脉瓣开放时血液从右心室流到肺。
新生儿的肺动脉瓣狭窄病情轻重不一,轻者不需要治疗,重者可能威胁生命。
大多数肺动脉瓣狭窄的小儿,瓣膜多是轻度到中度狭窄。肺动脉瓣狭窄时,右心室泵血困难,需要较大的力量才能让血流通过肺动脉瓣。如果有中度肺动脉瓣狭窄,体格检查、心电图、心动图、心导管检查可确诊。治疗可以通过大腿的静脉(股静脉)插入一个带气囊的倾斜的塑料管将瓣膜扩张。如果瓣膜发育不好,需要外科手术重建瓣膜。
如果肺动脉瓣狭窄非常严重,只有很少的静脉血流到肺循环进行气体交换,右心室、右心房压力继发性增加,静脉血通过左右房间隔,流到左心室、主动脉,然后到全身各个部位。这样,婴儿可能出现青紫――发绀。当出现发绀时,应给予前列腺素类药物如前列地尔以保持动脉导管开放,前列地尔一直使用到外科医生能在主动脉和肺动脉之间做搭桥手术,或手术扩张肺动脉瓣,或在某些病例二者都做。这样能使血液绕过狭窄的肺动脉瓣流到肺进行氧气交换。有些儿童成年后还需再做一次手术。
. 主动脉缩窄
主动脉缩窄是主动脉的一个狭窄,常常发生于主动脉和动脉导管连接处、降主动脉及腹腔动脉。
主动脉是一个大动脉血管,它携带动脉血从心脏到全身各个部位。主动脉缩窄导致下肢血流量减少,下肢的脉搏和血压较正常低,而上肢的脉搏和血压较正常高。在大多数病例,主动脉缩窄不会引起任何问题。有些儿童由于上肢血压高出现头痛和鼻出血,由于下肢血压低出现运动后下肢疼痛,但大多数无症状。主动脉缩窄的儿童常有主动脉瓣的异常,主动脉瓣只有二叶,正常主动脉瓣为三叶。
由于有脉搏和血压的变化,根据体格检查可以怀疑诊断,X线片、心电图、心动图可以确诊。主动脉缩窄时左心室需要更大的压力才能将血泵到缩窄的主动脉内,故应在儿童期及早手术以减少左心室的负荷,避免年长后出现高血压等并发症。一般手术在学龄前(3~5岁)完成。
主动脉缩窄的小儿在生后几天到2周,在动脉异管闭合后可能出现严重的心力衰竭,患儿呼吸困难,面色苍白。血液检测显示血中酸度明显增加(代谢性酸中毒)。这种情况严重威胁生命,需及时纠正酸中毒,并给予适当的治疗。治疗包括给予前列腺素类药物如前列地尔将动脉导管重新开放,给予支持心脏的药物,急诊手术。这种手术可以挽救新生儿的生命。有些儿童年长后还需再作一次手术。常常还有一些合并症如主动脉瓣二叶、主动脉狭窄、二尖瓣异常、室间隔缺损也需要治疗。
. 大血管异位
大血管异位是指主动脉和肺动脉与心脏的正常连接的错位,即主动脉与右心室相连,肺动脉与左心室相连。
正常情况下,肺动脉携带静脉血从右心室到肺,主动脉携带动脉血从左心室到全身。大血管异位时,从全身回到右心室的静脉血流到主动脉,主动脉又将静脉血带到全身,不经过肺循环。婴儿体内大量的动脉血只通过肺循环,而不流经全身的其他部位。
新生儿出生后可以暂时存活。正常情况下,出生时右心房和左心房之间有一个小孔(卵圆孔)存在,卵圆孔开放可以允许一部分血液从肺分流到左心房再到右心房,然后到右心室和主动脉,这样可以供给全身一定的氧气使婴儿能存活。
出生后,通过体格检查、X线片、心电图、心动图可以很快作出诊断。一般在生后几天内应进行手术治疗。手术过程包括将主动脉和肺动脉分别与左心室和右心室相连,冠状动脉再植。冠状动脉保障心脏自身的供血,在大血管异位的病人,冠状动脉可能异位。手术前,有些婴儿应给予前列腺素如前列地尔以保持动脉导管开放,有些病人需要用一个带气囊的倾斜的导管在扩大的动脉间开一个通道,允许更多的动脉血达到主动脉。
. 左室发育不良综合征
左室发育不良综合征又称左心发育不良综合征。左心室的基本功能是将血泵到全身。左心室和瓣膜严重发育不良或缺乏,不能保证全身的供血。出生时,由于血液能通过未闭合的动脉导管从右心室流到全身,婴儿不出现症状,一旦动脉导管闭合后,就会出现严重的心力衰竭。大多数患儿都不能存活。
. 法洛四联症
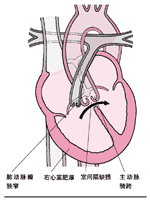
法洛四联症是大的室间隔缺损、主动脉骑跨(允许静脉血直接从右心室流到主动脉)、肺动脉口狭窄(心脏右侧的流出道狭窄)、右心室肥厚组成的一组心脏畸形。
法洛四联症的婴儿出生时或出生后不久常常可以听到心脏杂音。全身循环的血液由于携氧不足而出现发绀。由于肺动脉口狭窄限制了肺的血流量,右心室含氧少的静脉血通过室间隔缺损到左心室,再到主动脉,然后循环到全身。有些患儿有轻度发绀,且不加重,手术可以等到婴儿后期再作;有些患儿有严重的症状,影响了正常的生长发育;有些患儿可能有突然发作或间断发作发绀,活动后如哭闹或便后发绀加重。发绀的婴儿可能呼吸短促,或意识丧失。如果婴儿有间断发作的发绀,应输氧和给予吗啡治疗,然后临时性地给予心得安以防再次发作。这种婴儿需要及时手术治疗,手术修复法洛四联症,或在主动脉和肺动脉之间造一个临时性的人工通道以增加肺的血流量,进行气体交换。
|