á161ñTRANSFUSION AND INFUSION ASSEMBLIES AND SIMILAR MEDICAL DEVICES
The requirements apply to sterile and nonpyrogenic assemblies or devices in contact directly or indirectly with the cardiovascular system,the lymphatic system,or cerebrospinal fluid.This includes,but is not limited to,solution administration sets,extension sets,transfer sets,blood administration sets,intravenous catheters,implants extracorporeal oxygenator tubings and accessories,dialysers and dialysis tubing and accessories,heart valves,vascular grafts,intramuscular drug delivery catheters,and transfusion and infusion assemblies.These requirements do not apply to orthopedic products,latex gloves,or wound dressings.
Bacterial Endotoxins— Proceed as directed under Bacterial Endotoxins Test á85ñ.
For medical devices,the endotoxin limit is not more than 20.0USP Endotoxin Units per device except that for those medical devices in contact with the cerebrospinal fluid the limit is not more than 2.15USP Endotoxin Units per device.
Adevice that fails this test can be retested once by another Bacterial Endotoxinstest.For devices that cannot be tested by the Bacterial Endotoxins Test á85ñbecause of nonremovable inhibition or enhancement,the Pyrogen Test á151ñis applied.
Preparation of Devices—
Select not less than 3and not more than 10devices.Rinse or soak the devices with LAL Reagent Water.The volume of rinsing or extracting solution may be adjusted for the size and configuration of the device.
For devices labeled “nonpyrogenic fluid pathway,”flush the fluid pathway with extracting fluid that has been heated to 37±1.0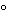 ,keeping the extracting fluid in contact with the relevant pathway for not less than 1hour at controlled room temperature.Extracts may be combined,where appropriate.The endotoxin limit for the rinsing or extracting solution is calculated by the formula:
,keeping the extracting fluid in contact with the relevant pathway for not less than 1hour at controlled room temperature.Extracts may be combined,where appropriate.The endotoxin limit for the rinsing or extracting solution is calculated by the formula:
(K×N)/(V),
where Kis equal to the amount of endotoxin allowed per device,Nis equal to the number of devices tested,and Vis equal to the total volume of the extract or rinse.If the undiluted rinsing or extracting solution is unsuitable for the Bacterial Endotoxins Test á85ñ,repeat the inhibition or enhancement test after neutralization and removal of the interfering substances or after the solution has been diluted by a factor not exceeding the Maximum Valid Dilution.The Maximum Valid Dilution for devices is calculated by dividing the endotoxin limit by the labeled sensitivity lof the LALreagent used.
Pyrogen— For samples that cannot be tested by the Bacterial Endotoxins Testbecause of nonremovable inhibition or enhancement of the test,the Pyrogen Test á151ñis applied.Select 10devices,and obtain a pooled effluent,utilizing preparation methods appropriate to the device as directed for Bacterial Endotoxins,but with volumes of rinse or extraction fluid not to exceed 40mLof sterile saline TSper device.The requirements of the Pyrogen Test á151ñare met.
Other Requirements— The portions of medical devices that are made of plastics or other polymers meet the requirements specified for Biological Tests—Plastics and Other Polymersunder Containers á661ñ;those made of elastomers meet the requirements under Elastomeric Closures for Injections á381ñ.If a class designation for elastomers,plastics,or other polymers is needed,perform the appropriate in vivo tests indicated in the general test chapter Biological Reactivity Tests,In Vivo á88ñ.