Famotidine Tablets
»Famotidine Tablets contain not less than 90.0percent and not more than 110.0percent of the labeled amount of famotidine (C8H15N7O2S3).
Packaging and storage—
Preserve in well-closed,light-resistant containers.Store at controlled room temperature.
Identification—
A:
(See Thin-Layer Chromatographic Identification Test á201ñ.)
Developing solvent—
Prepare a mixture of ethyl acetate,methanol,toluene,and ammonium hydroxide (40:25:20:2).
Standard solution—
Dissolve USP Famotidine RSin glacial acetic acid to obtain a solution having a concentration of 4mg per mL.
Test solution—
Transfer a portion of finely powdered Tablets,equivalent to about 40mg of famotidine,to a 10-mLvolumetric flask.Dissolve in glacial acetic acid with the aid of sonication,dilute with glacial acetic acid to volume,and centrifuge to get a clear liquid.
Procedure—
Apply separately 10µLeach of the Standard solutionand the Test solutionto a suitable thin-layer chromatographic plate coated with a 0.25-mm layer of chromatographic silica gel mixture,allow the spots to dry,and develop the plate in a paper-lined chromatographic chamber equilibrated with Developing solventfor about 1hour prior to use.Allow the chromatogram to develop until the solvent front has moved about 15cm.Remove the plate,air-dry,and examine the plate under short-wavelength UVlight:the principal spot from the Test solutioncorresponds in appearance and RFvalue to that of the Standard solution.
B:
The retention time of the major peak in the chromatogram of the Assay preparationcorresponds to that in the chromatogram of the Standard preparation,as obtained in the Assay.
Dissolution á711ñ—
Medium:
pH4.5,0.1Mphosphate buffer;prepared by dissolving 13.6g of monobasic potassium phosphate in 1Lof water;900mL.
Apparatus 2:
50rpm.
Time:
30minutes.
Procedure—
Determine the amount of C8H15N7O2S3dissolved from UVabsorption at the wavelength of maximum absorbance at about 265nm,using filtered portions of the solution under test,suitably diluted with Mediumif necessary,in comparison with a Standard solution having a known concentration of USP Famotidine RSin the same Medium.
Tolerances—
Not less than 75%(Q)of the labeled amount of C8H15N7O2S3is dissolved in 30minutes.
Related compounds—
Buffer solution,Mobile phase,Diluent,System suitability solution,Standard preparation,Assay preparation,and Chromatographic system—
Proceed as directed in the Assay.
Standard solution—
Use the Standard preparation.
Test solution—
Use the Assay preparation.
Procedure—
Separately inject a volume (about 50µL)of the Test solutioninto the chromatograph,record the chromatograms,and measure the responses for the major peaks.Calculate the percentage of each impurity in the portion of Tablets taken by the formula:
In addition to not exceeding the limits for each impurity in Table 1,not more than 1.5%of total impurities is found.
100(1/F)C(D/LN)(ri/rs),
in which Fis the relative response factor for each impurity peak (see Table 1for values);Cis the concentration,in mg per mL,of USP Famotidine RSin the Standard solution;Lis the labeled amount,in mg,of famotidine in each Tablet;Nis the number of tablets taken to prepare the Test solution;Dis the dilution factor used to prepare the Test solution;riis the peak area obtained for each individual impurity in the Test solution;and rsis the peak area for famotidine in the Standard solution.
Table 1
| Relative Retention Time |
Relative Response Factor (F) |
Name | Limit (%) |
| 0.38 | 1.0 | Famotidine related com pound A1 | 1.0 |
| 0.65 | 1.0 | Famotidine related com pound B2 | 0.5 |
| 0.85 | 1.0 | Famotidine related com pound C3 | 0.5 |
| 1.21 | 1.3 | Famotidine related com pound D4 | 0.5 |
|
1
3-[2-(diaminomethyleneamino)-1,3-thiazol-4-ylmethylsulfinyl]-N-sulfamoyl-propanamidine
|
|||
|
2
3-[2-(diaminomethyleneamino)-1,3-thiazol-4-ylmethylthio]-propanoic acid
|
|||
|
3
3-[2-(diaminomethyleneamino)-1,3-thiazol-4-ylmethylthio]-N-sulfamoyl-propanamide
|
|||
|
4
3-[2-(diaminomethyleneamino)-1,3-thiazol-4-ylmethylthio]-propanamide
|
|||
Uniformity of dosage units á905ñ:
meet the requirements.
Assay—
Buffer solution—
Dissolve 13.6g of sodium acetate trihydrate in 750mLof water.Add 1mLof triethylamine,adjust with glacial acetic acid to a pHof 6.0,and dilute with water to 1L.
Mobile phase—
Prepare a mixture of Buffer solutionand acetonitrile (93:7),mix,and degas.Make adjustments if necessary (see System Suitabilityunder Chromatography á621ñ).
Diluent—
Dissolve 6.8g of monobasic potassium phosphate in 750mLof water,adjust with 1Mpotassium hydroxide to a pHof 6.0,and dilute with water to 1L.
System suitability stock solution—
Transfer 10mg of famotidine to a 50-mLvolumetric flask,add 1mLof 0.1Nhydrochloric acid,heat at 80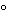 for 30minutes,and cool to room temperature.Add 2mLof 0.1Nsodium hydroxide,heat at 80for 30minutes,cool to room temperature,and neutralize by adding 1mLof 0.1Nhydrochloric acid.Dilute with Diluentto volume.Transfer 10mLof this solution to a separate 50-mLvolumetric flask containing 5mg of famotidine dissolved in 8mLof methanol.Dilute with Diluentto volume.
for 30minutes,and cool to room temperature.Add 2mLof 0.1Nsodium hydroxide,heat at 80for 30minutes,cool to room temperature,and neutralize by adding 1mLof 0.1Nhydrochloric acid.Dilute with Diluentto volume.Transfer 10mLof this solution to a separate 50-mLvolumetric flask containing 5mg of famotidine dissolved in 8mLof methanol.Dilute with Diluentto volume.
System suitability solution—
Transfer about 1mLof System suitability stock solution to a suitable container,add 1mLofDiluentand 1drop of hydrogen peroxide solution,and mix well.[NOTE—Prepare fresh daily.]
Standard preparation—
Transfer about 10mg of USP Famotidine RS,accurately weighed,into a 100-mLvolumetric flask,add 20mLof methanol,and sonicate for 5minutes.Dilute with Diluentto volume,and mix.
Assay preparation—
Transfer not fewer than 10Tablets to a 1-Lvolumetric flask.Add 200mLof Diluent,and swirl to erode the Tablets.Add 200mLof methanol,and stir by mechanical means at 300rpm for 1hour.Dilute with Diluentto volume,mix,and filter.Quantitatively dilute a portion of the clear filtrate with Diluentto obtain a solution containing about 0.1mg of famotidine per mL.
Chromatographic system (see Chromatography á621ñ)—
The liquid chromatograph is equipped with a 275-nm detector and a 4.6-mm ×15-cm column that contains packing L1.The column temperature is maintained at 40.The flow rate is about 1.4mLper minute.Chromatograph the System suitability solution,and identify the famotidine peak and the peaks due to impurities and degradation products listed in Table 1.Record the peak responses as directed for Procedure:the resolution,R,between famotidine related compound Cand famotidine is not less than 1.3;the resolution,R,between famotidine and famotidine related compound Dpeaks is not less than 1.3;and the capacity factor,k¢,for famotidine peak is not less than 2.0.Chromatograph the Standard preparation,and record the peak responses as directed for Procedure:the relative standard deviation for replicate injections is less than 2.0%.
Procedure—
Separately inject equal volumes (about 50µL)of the Standard preparationand the Assay preparationinto the chromatograph,record the chromatograms,and measure the responses for the major peaks.Calculate the quantity,in mg,of famotidine (C8H15N7O2S3)in each Tablet taken by the formula:
C(D/N)(rU/rS),
in whichCis the concentration,in mg per mL,of USP Famotidine RSin the Standard preparation;Dis the dilution factor used to prepare the Assay preparation;Nis the number of Tablets taken to prepare the Assay preparation;and rUand rSare the peak areas obtained from the Assay preparationand the Standard preparation,respectively.
Auxiliary Information—
Staff Liaison:Elena Gonikberg,Ph.D.,Scientist
Expert Committee:(PA4)Pharmaceutical Analysis 4
USP28–NF23Page 806
Pharmacopeial Forum:Volume No.29(3)Page 627
Phone Number:1-301-816-8251